
搜索结果: 1-8 共查到“材料科学 理论计算”相关记录8条 . 查询时间(0.267 秒)
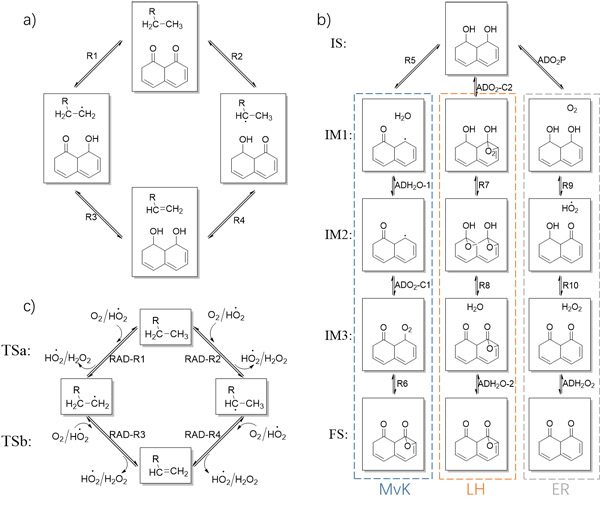
烷烃催化脱氢反应过程理论计算模拟取得新进展(图)
烷烃 催化 脱氢反应 低碳烯烃
2020/11/18
低碳烯烃是化工产业的支柱,是合成塑料、橡胶和纤维的基本原材料。烯烃产量是衡量一个国家化工产业能力重要指标之一,随着经济发展烯烃需求在持续增加,2020年我国烯烃消耗量占全球15%以上。由此可见,进一步提高烯烃生产效率有着重要经济价值和社会意义。另一方面,通过烷烃催化脱氢反应可以高效将低碳烷烃分子转化为同碳烯烃。目前,烷烃催化脱氢(直接和氧化)反应面临着选择性低、积碳以及高能耗等挑战,设计和开发新型...
分子间C-F…H-C 赝氢键电子结构与性质的理论计算
弱相互作用 赝氢键 电子结构 电子密度拓扑性质
2012/3/22
采用密度泛函B3LYP (Becke, three-parameter, Lee-Yang-Parr)/6-311++G**和HF(HartreeFock)/6-311++G**方法, 从理论上探讨了2-F-四氢呋喃(2-F-Tetrahydrofuran)分别与几种常见而重要的生物小分子咪唑(Iminazole)、嘧啶(Pyrimidine)、腺嘌呤(Adenine)和鸟嘌呤(Guanine)等...
双剪统一强度理论计算塑性金属材料强度的唯一性
双剪统一强度理论 屈服 塑性 金属
2009/8/26
研究了双剪统一强度理论及其有关关系式,得到塑性金属材料双剪统一强度理论参数b的计算式,对某一塑性金属材料,其值为常数,取值范围是b>−1;运用双剪统一强度理论计算某一塑性金属材料的强度时有唯一确定的强度计算值,而不是多个值,不能得出塑性金属材料的τs/σs;分析材料的拉压强度比为1时双剪统一强度理论的2组等价变换式,对b<0的材料,双剪统一强度理论计算表明,材料的破坏不是由中间主剪应力...
高孔率金属延伸率的理论计算
高孔率金属 延伸率 计算 孔率
2008/11/20
运用几何学与力学推导各同性高孔率材料的孔率与延伸率的近似关系. 通过对高孔隙率发泡镍的实验, 证明了理论公式与实际结果一致.
Fe--Mn--Si合金相变热滞的理论计算
Fe--Mn--Si合金 相变 热滞(As-Ms)
2007/11/8
文章摘要:
利用相变热力学和界面摩擦概念, 推导出Fe--Mn--Si合金相变热滞(As-Ms)的一般计算表达式; 根据此关系式, 计算了这种合金的相变热滞, 计算结果与实验值相符. 合金的界面摩擦能耗比较大, 且与合金的热滞成正比, 由此可知合金的热滞大是由于界面摩擦能耗大所造成的.
有效介质理论计算He原子在金属钒中的扩散行为
钒 He原子 有效介质理论
2007/11/8
采用有效介质理论, 并考虑晶格驰豫的影响计算He原子在金属钒中的嵌入能, 通过能量分析推测He原子在金属钒中可能的扩散路径和扩散势垒. 计算结果表明, He原子在金属钒bcc结构的四面体间隙位置有能量的最低点, 为4.37 eV, 在八面体间隙位置嵌入能比四面体间隙位置的稍大, He原子在金属钒的bcc结构中最有可能在(100)面内沿着由位置(1, 1/4, 1/2), (1, 1/2, 1/2)...
多硝基[60]富勒烯衍生物C-N键离解能的理论计算
物理化学 多硝基[60]富勒烯 键离解能 平均键离 解能 硝基平均结合能
2007/3/17
用PBE密度泛函方法计算了14种多硝基[60]富勒烯的最弱 C-N键离解能、C-N键平均键离解能以及硝基的平均结合能。计算结果表明,最弱C-N键的离 解能为137.14~158.51kJ/mol,C-N键的平均键离解能为168.56~177.78kJ/mol,硝基平均结合能为-40.47~-50.92k J/mol。硝基数从2个增加到8个,键离解能的改变不大,但1,2连接模式优于1,4连接模式, ...